...
- In several tutorials (MultiQC, fastp, Advanced Breseq) I gave example command line for loops that can be used to generate commands files for large numbers of samples.
- Often I use a python script to generate files like this, as I am more fluent in python coding than I am in bash/command line scripting.
- Sometimes I generate some commands in Excel
- An example might be a formula of
'="breseq -j 6 -p -r Ref.gbk -o Run_output/" & A1 & " " & B1 & " " & C1 & " >& runLogs/" & A1'with sample name in A1, read1 in B1 and read2 in C1.
- An example might be a formula of
- In a program like BBedit/notepad to copy paste the same command multiple times and change it as appropriate then paste the text directly into a nano editor on TACC.
- In the introduction tutorial, several alternative text editor programs were mentioned with different ssh connectivity
...
- The ability to have a lab wide conda environment file or share a conda environment with other people in your lab to make sure that everyone is working from the same starting point and with tools that should work. I can tell you a sizable portion of the time I spend in lab helping other members is devoted to dealing with accessing programs. I look forward Being able to just being able to share a conda environment file with them significantly improves this.
- In the section dealing with selecting tools, I talk a bit about publishing, and how some papers struggle to define exactly what they did computationally with enough accuracy detail to be able to make use of the analysis they did in looking at my own work. Conda environments are increasingly being shared as part of supplemental information not just in/for publications about computational based programs, but for any paper involving command line analysis. I hope that this trend continues, and that you see the value in doing such things and contribute to it.
- As mentioned early in class, your laptop is not going to be capable of performing the majority of the analysis you are going to want to do. That being said, sometimes we have to work with what we have, or it can be useful to look at or build things on your local machine before transferring the analysis to TACC. Downstream analysis often requires working with python/R/etc to generate figures for publication. Putting required visualization packages into a single environment (or given number of environments) can aid in publication reproducabilityreproducibility/interaction as mentioned above.
- Other reasons! I haven't been working with conda in my own work enough yet to have a complete feel for all the ways it helps things (and much of this is based around this ability to transfer environments. I would not be surprised to hear from you that you have thought of another way it can be helpful in your own work.
| Code Block | ||||
|---|---|---|---|---|
| ||||
cdh for pathway in $(for conda_env in ~/miniconda3/envs/*; `conda env list`;do echo $conda_env|grep "^/.*miniconda3\/envs";done); do env_name=$(echo $conda_env$pathway|sed 's/.*\///');echo $env_name;conda activate $env_naamename;conda list;done > list_of_envrionments_used_in_GVA2021GVA2022_and_packages_installed.txt # for pathway in $(for conda_env in ~/miniconda3/envs/*`conda env list`;do echo $conda_env|grep "^/.*miniconda3\/envs";done); do env_name=$(echo $conda_env$pathway|sed 's/.*\///');echo $env_name;conda activate $env_naamename;conda env export > $env_name.yml;done # mkdir GVA2021GVA2022_conda_files mv *.yml GVA2021GVA2022_conda_files mv list_of_envrionments_used_in_GVA2021GVA2022_and_packages_installed.txt GVA2021GVA2022_conda_files tar -czvf GVA2021GVA2022_conda_files.tar.gz GVA2021GVA2022_conda_files/ |
Using the scp tutorial you can now transfer the GVA2021the GVA2022_conda_files.tar.gz file back to your computer
...
- When you type a command, only locations that are in your PATH variable are searched for an executable command matching that name.
- When the command is found in your PATH, the computer immediately substitutes the location it was found for the short command name you entered, and stops searching.
- This means that things that are early in your path are always searched first. In some extreme circumstances if you add a bunch of locations with lots of files to the front of your PATH, you can actually slow down your entire computer, so try to limit the path variable to only look in directories containing executable files.
- You can manually manipulate your $PATH variable, usually in your .bashrc file in your home direcotrydirectory.
Warning title One of the most important lessons you can ever learn Anytime you manipulate your PATH variable you always want to make sure that you include $PATH on the right side of the equation somewhere separated by : either before it, after it, or on both sides of it if you want it in the middle of 2 different locations. As we are explaining right now, there are reasons and times to put it in different relative places, but if you fail to include it (or include a typo in it by calling it say $PTAH) you can actually remove access to all existing commands including the most basic things like "ls" "mkdir" "cd".
...
- It is not widely used/downloaded.
- Even I consider myself a novice at best when it comes to computational analysis. I rely on others who are more versed in the computer science, biostatistics, etc to evaluate tools.
- If you try to publish using a tool that is not common to the field or worse that none of the reviewers have ever heard of, you will deal with it in the review process, including potentially having to redo analysis with better accepted/common tools as a condition of publication
- Difficult to install
- Lack example command on command line to model your work after
- Lack command line help detailing what options are expected/available
- Present in a paper but no github/website/forum.
- Broadly this suggests not actively being developed, and increasing liklihood likelihood you will have difficulties getting answers to questions or help if there are problems.
...
- Any version that works to get you closer to answering the biological question that does not have a newer version that "fixes bugs/errors associated with" the version you want to use .for the options you are selecting.
- Most new versions deal with expanding features, or some kind of speed/memory improvement. Very few new versions result in producing different results.
- Generally the bigger the value change, the bigger the change to the program. Changing from version 1 to version 2 is a very big change. version 1.1 to 1.2 less big 1.15 1.5 to 1.1.16 6 smaller still etc. There is a lot of wiggle room in this rule.
- Version similar to that which others are publishing about.
- If field is publishing using gatk version 4.0 and you have to cite gatk version 2.0 those are very different.
- A single version for all analysis in a single paper, or at least section of the methods.
- Personally I am incredibly frustrated with papers that say something like "versions x to y of toy Z were used". This is going to make it very hard to figure out how to mimic their analysis, repeat their analysis, evaluate their analysis.
- Not listing a version of a computational program is even worse.
- Not citing a paper as tools request is worse still.
- The bad thigns things listed above are also associated with papers that do not detail what actual commands were used in analysis, again making it harder to mimic their analysis.
- If you do decide to update the version of a program, repeat the analysis you have already done.
- You should be conducting your analysis in such a way that you can repeat the analysis for exactly this reason.
- Personally I am incredibly frustrated with papers that say something like "versions x to y of toy Z were used". This is going to make it very hard to figure out how to mimic their analysis, repeat their analysis, evaluate their analysis.
...
- Results you have don't make sense, can't be validated, or are inconsistent.
- A major bug effecting analysis is repaired.
- can Can be difficult to determine what the effect a version change will have. If the documentation calls it a "major bug" or describes something that impacts your analysis.
- forums Forums of some of the better programs will break down what change changes a little better and most will respond to any inquiries of "do i I need to update".
- If looking for help on an error or problem with the program and lots of answers deal with mentioning specific versions being wrong/bad/the source of the problem and you are using that version (even if you don't think you are having problems).
- An existing pipeline no longer works with new sample/data/improvement.
- Has new feature you want to try.
- Probably a good idea to test this in separate environment.
- Staring work on a new project/paper or new analysis in an on going project/paper that uses the same tool.
Transferring files
The majority of the files we have worked with have been in our $SCRATCH space. Recall that files on $SCRATCH can be deleted after a period of inactivity. Below is a list of things that you SHOULD copy to your $HOME or $WORK2 $WORK space.
Collecting class information via job submission
commands
Navigate to the $SCRATCH directory before doing the following.
...
| Code Block | ||||||
|---|---|---|---|---|---|---|
| ||||||
echo "My name is _____ and todays date is:" > GVA2021GVA2022.output.txt date >> GVA2021GVA2022.output.txt echo "I have just demonstrated that I know how to redirect output to a new file, and to append things to an already created file. Or at least thats what I think I did" >> GVA2021GVA2022.output.txt echo "i'm going to test this by counting the number of lines in the file that I am writing to. So if the next line reads 4 I remember I'm on the right track" >> GVA2021GVA2022.output.txt wc -l GVA2021GVA2022.output.txt >> GVA2021GVA2022.output.txt echo "I know that normally i would be typing commands on each line of this file, that would be executed on a compute node instead of the head node so that my programs run faster, in parallel, and do not slow down others or risk my tacc account being locked out" >> GVA2021GVA2022.output.txt echo "i'm currently in my scratch directory on stampede2. there are 2 main ways of getting here: cds and cd $SCRATCH" >>GVA2021>>GVA2022.output.txt pwd >> GVA2021GVA2022.output.txt echo "over the last week I've conducted multiple different types of analysis on a variety of sample types and under different conditions. Each of the exercises was taken from the website https://wikis.utexas.edu/display/bioiteam/Genome+Variant+Analysis+Course+20212022" >> GVA2021GVA2022.output.txt echo "using the ls command i'm now going to try to remind you (my future self) of what tutorials I did" >> GVA2021GVA2022.output.txt ls -1 >> GVA2021GVA2022.output.txt echo "the contents of those directories (representing the data i downloaded and the work i did) are as follows: ">> GVA2021GVA2022.output.txt find . >> GVA2021GVA2022.output.txt echo "the commands that i have run on the headnode are: " >> GVA2021GVA2022.output.txt history >> GVA2021GVA2022.output.txt echo "the contents of this, my commands file are: ">>GVA2021>>GVA2022.output.txt cat commands >> GVA2021GVA2022.output.txt echo "I will next creatcreate a what_i_did_at_GVA2021GVA2022.slurm file that will run for 15 minutes" >> GVA2021GVA2022.output.txt echo "and i will send this job to the queue using the the command: sbatch what_i_did_at_GVA2021GVA2022.slurm" >> GVA2021GVA2022.output.txt |
| Code Block | ||||||
|---|---|---|---|---|---|---|
| cp /corral-
| |||||
wc -l commands |
If you get a number larger than 19 edit your commands file with nano so each command is a single line as they appear above. Several of the lines are likely long enough that they will wrap when you paste them in nano and cause problems
| Code Block | ||||
|---|---|---|---|---|
| ||||
cp /corral-repl/utexas/BioITeam/gva_course/GVA2021GVA2022.launcher.slurm what_i_did_at_GVA2021GVA2022.slurm nano what_i_did_at_GVA2021GVA2022.slurm |
As stated above things we want to change are:
| Line number | As is | To be |
|---|---|---|
| 16 | #SBATCH -J jobName | #SBATCH -J end_of_class |
| 21 | #SBATCH -t 12:00:00 | #SBATCH -t 0:10:00 |
| 27 | conda activate GVA2021 | conda activate GVA2021 |
Line 27 could be deleted if you prefer.
Again use ctl-o and ctl-Again use ctl-o and ctl-x to save the file and exit.
...
| Code Block | ||||
|---|---|---|---|---|
| ||||
sbatch what_i_did_at_GVA2021GVA2022.slurm |
| Code Block | ||||
|---|---|---|---|---|
| ||||
wc -l commands |
If you get a number larger than 19 edit your commands file with nano so each command is a single line as they appear above. Several of the lines are likely long enough that they will wrap when you paste them in nano and cause problems
Evaluating your job submission
...
Evaluating your job submission
Based on our example you may have expected 1 new file to have been created during the job submission (GVA2022.output.txt), but instead you will find 2 extra files as follows: what_i_did_at_GVA2020GVA2022.e(job-ID), and what_i_did_at_GVA2020GVA2022.o(job-ID). When things have worked well, these files are typically ignored. When your job fails, these files offer insight into the why so you can fix things and resubmit.
...
| Code Block | ||||||
|---|---|---|---|---|---|---|
| ||||||
# remember that things after the # sign are ignored by bash cat GVA2021GVA2022.output.txt > end_of_class_job_submission.final.output mkdir $WORK2$WORK/GVA2021GVA2022 mkdir $WORK2$WORK/GVA2021/end_of_course_summary/ # each directory must be made in order to avoid getting a no such file or directory error cp end_of_class_job_submission.final.output $WORK2$WORK/GVA2021GVA2022/end_of_course_summary/ cp what_i_did* $WORK2$WORK/GVA2021/end_of_course_summary/ # note this grabs the 2 output files generated by tacc about your job run as well as the .slurm file you created to tell it how to run your commands file cp commands $WORK2/GVA2021/end_of_course_summary/GVA2022/end_of_course_summary/ # note this grabs the 2 output files generated by tacc about your job run as well as the .slurm file you created to tell it how to run your commands file cp commands $WORK/GVA2022/end_of_course_summary/ |
Copy paste alternative to transfer
As we have seen several times in class, many output files are comma, or space, or tab delimited . While this type of formatting is often required for downstream applications, it can make it very difficult to look at if you are just trying to get a feel for what is going on. One solution to this is often to copy from the terminal screen to Excel. Sometimes Excel will recognize the delimiter character and each chunk of data will go into its own Excell cell. Other times each line goes in column A (or worse the entire copy paste goes into cell A1. For reasons unknown to me, using the text editor BBEdit (formerly known as text wrangler, not to be confused with any association to TACC's naming conventions) as an intermediate can automatically convert tab (and some space) delimited text to the invisible tab characters Excel expects, and if not (or if it uses comma delimiters), BBEdit's find/replace interface is capable of working with the same regular expressions you are increasingly becoming familiar with from the command line programs (sed, grep, awk). While there is a paid version of BBEdit I have never even bothered using their free 30 day trial.
Additional use for BBEdit
BBEdit is also a very lightweight plain text editor. This means that it is capable of opening large files (such as genbank references) without huge amounts of buffering or "destination" formatting issues that something like microsoft word would have. https://www.barebones.com/products/bbedit/. If in your own work you find an alternative that seems to do much of the same functionality as described here, I would love to hear about it.
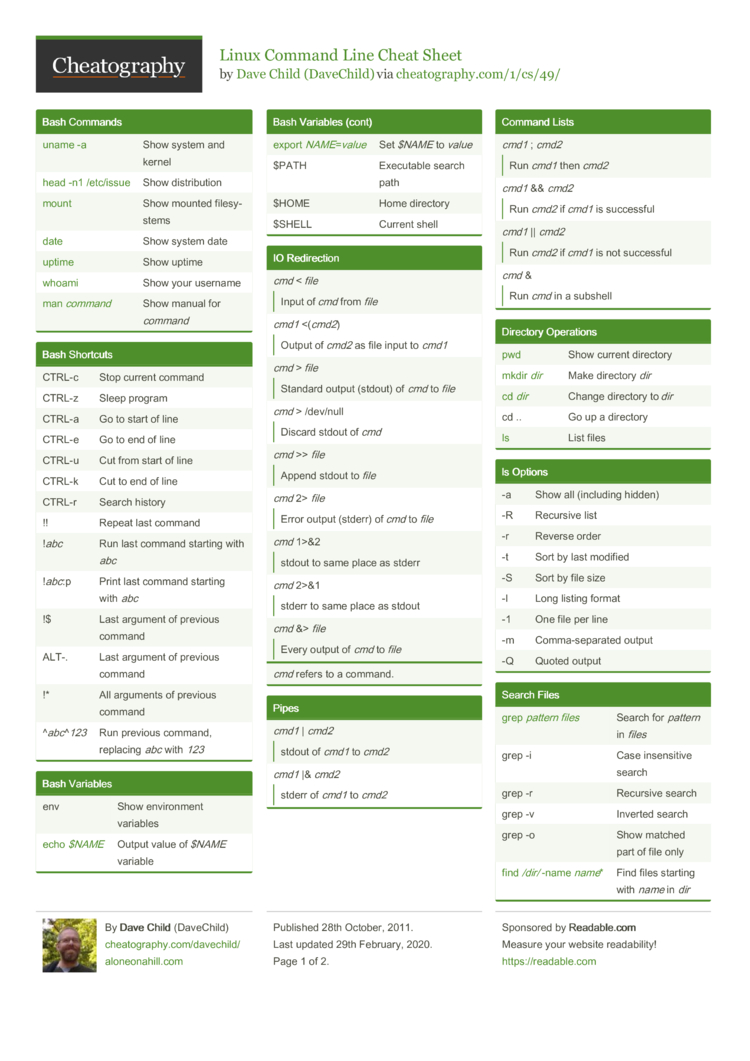
Helpful cheat sheets
cheat sheets are a common thing produced by people for common commands they want to use but don't always remember the exact formatting for or needed options. Here are a list of cheat sheets that may be helpful and what they are helpful for. They should at least provide you with the scope different cheat sheets are produced, if you find one lacking there are others that may have something you are looking for and can be found pretty quickly using google.
- For linux command line
- For conda:
{kind=link}
Return to GVA2021 GVA2022 to work on any additional tutorials you are interested in.